Contents
Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis is also known as cryptogenic fibrosing alveolitis and is a rare disease of unknown cause in which the lung becomes fibrotic. The average age at onset is 50 years although the age range is wide. The incidence is around 2 per 100,000 per year and the disease is slightly more common in females.
Idiopathic pulmonary fibrosis must be distinguished from other causes of pulmonary fibrosis, such as the following.
- Tuberculosis
- Sarcoidosis
- Systemic lupus erythematosis
- Rheumatoid arthritis
- Scleroderma
- Extrinsic allergic alveolitis
- Radiation
- Drugs (amiodarone, nitrofurantoin, bleomycin)
- Asbestos
- Inorganic dusts (pneunoconiosis, silicosis, berylliosis)
- Renal tubular acidosis (rare)
Pathology
Pulmonary fibrosis is classed as an interstitial lung disease. The fibrotic airways are stiff, thickened and difficult to open. In terms of
lung function tests, pulmonary fibrosis shows a reduction in all lung volumes. The FEV1/FVC ratio is preserved because the two volumes decrease in parallel.
The thickening of the walls of the alveoli increases the distance that oxygen and carbon dioxide have to diffuse to move between the blood and the air in the alveoli. This impairs gas exchange. The greater diffusing capacity of carbon dioxide means that hypoxia develops earlier in the disease than hypercapnia.
The basic process underlying pulmonary fibrosis is some form of injury to the alveolar walls. In idiopathic pulmonary fibrosis the nature of this injury remains obscure. The injury provokes an inflammatory response which first features oedema and the formation of hyaline membranes. The flat type one pneumocytes are replaced by the cuboidal type two pneumocytes. After this initial stage the second phase features a chronic inflammatory cell infiltrate in the walls of the alveoli. The cytokines released by these inflammatory cells induce the frequent partner of chronic inflammation: fibrosis. At first the fibrotic response consists of proliferation of fibroblasts (stage three) in preparation to lay down the fibrous tissue. The establishment of fibrosis and the accompanying architectural distortion is the fourth and final stage.
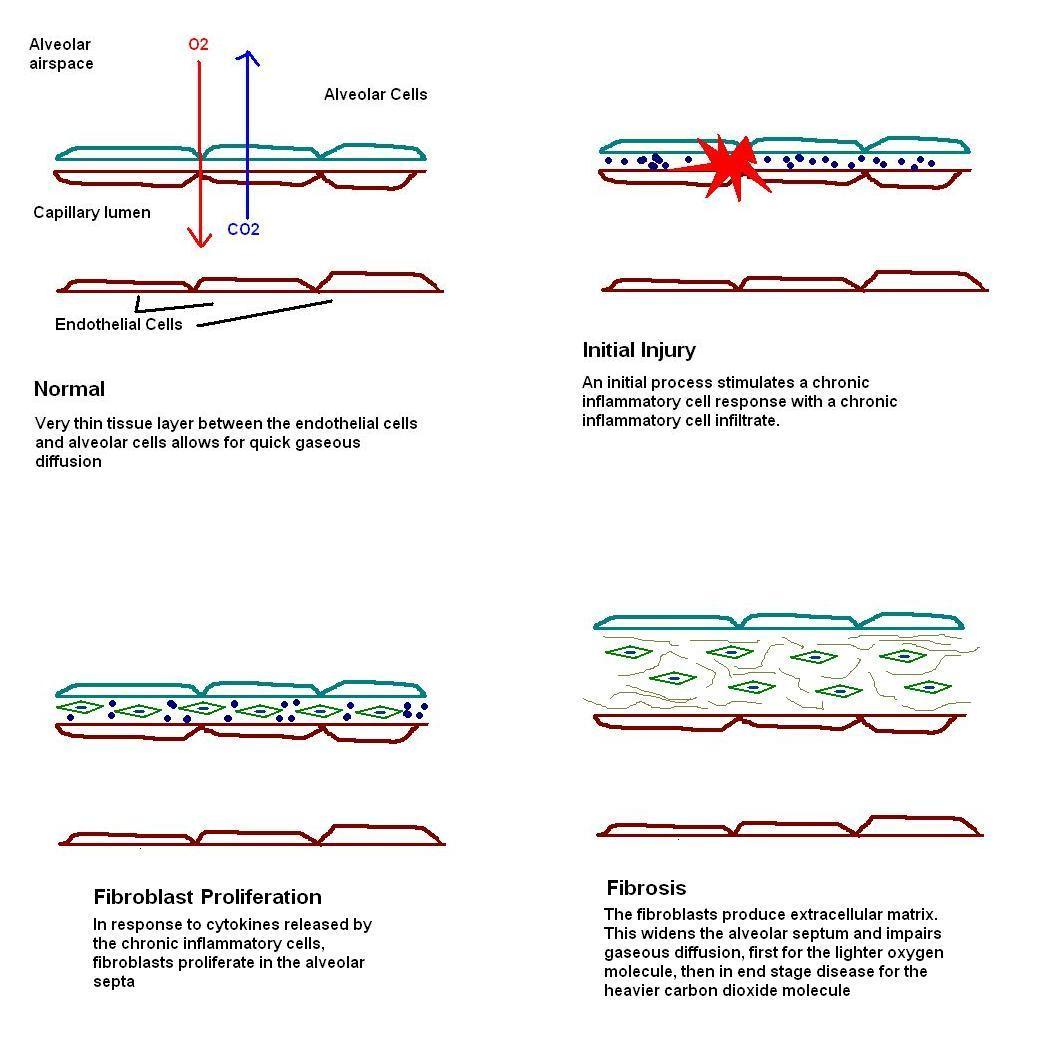
|
|
Diagram of the development of fibrosing alveolitis
|
Two main patterns of disease exist in idiopathic pulmonary fibrosis. Usual interstitial pneumonia is patchy and the lung typically has lesions at different stages of evolution. The changes are those of fibrosis and chronic inflammation. Desquamative interstitial pneumonia is dominated by the presence of numerous macrophages within the alveoli while the walls actually display only limited fibrosis; desquamative interstitial pneumonia is a more diffuse process.
Macroscopically the affected lungs gradually develop a honeycomb appearance due to the distortion of the airspaces. A fully developed honeycomb lung denotes end stage disease.

|
|
A lung which displays the end stage honeycomb pattern of pulmonary fibrosis
|
Clinical Features
Pulmonary fibrosis presents with a dry cough and progressive dyspnoea.
On examination patients may have clubbing. The trachea is central. Expansion is reduced because the airways are stiff and do not open properly. The percussion note may be dull due to the reduction in the volume of air in the lungs. The breath sounds remain vesicular but crackles are added. The vocal resonance may be increased as a consequence of the greater quantity of solid tissue.
In late disease pulmonary hypertension and right heart failure can develop.
Investigations
The purpose of the investigations is to confirm the presence of pulmonary fibrosis and to exclude secondary causes.
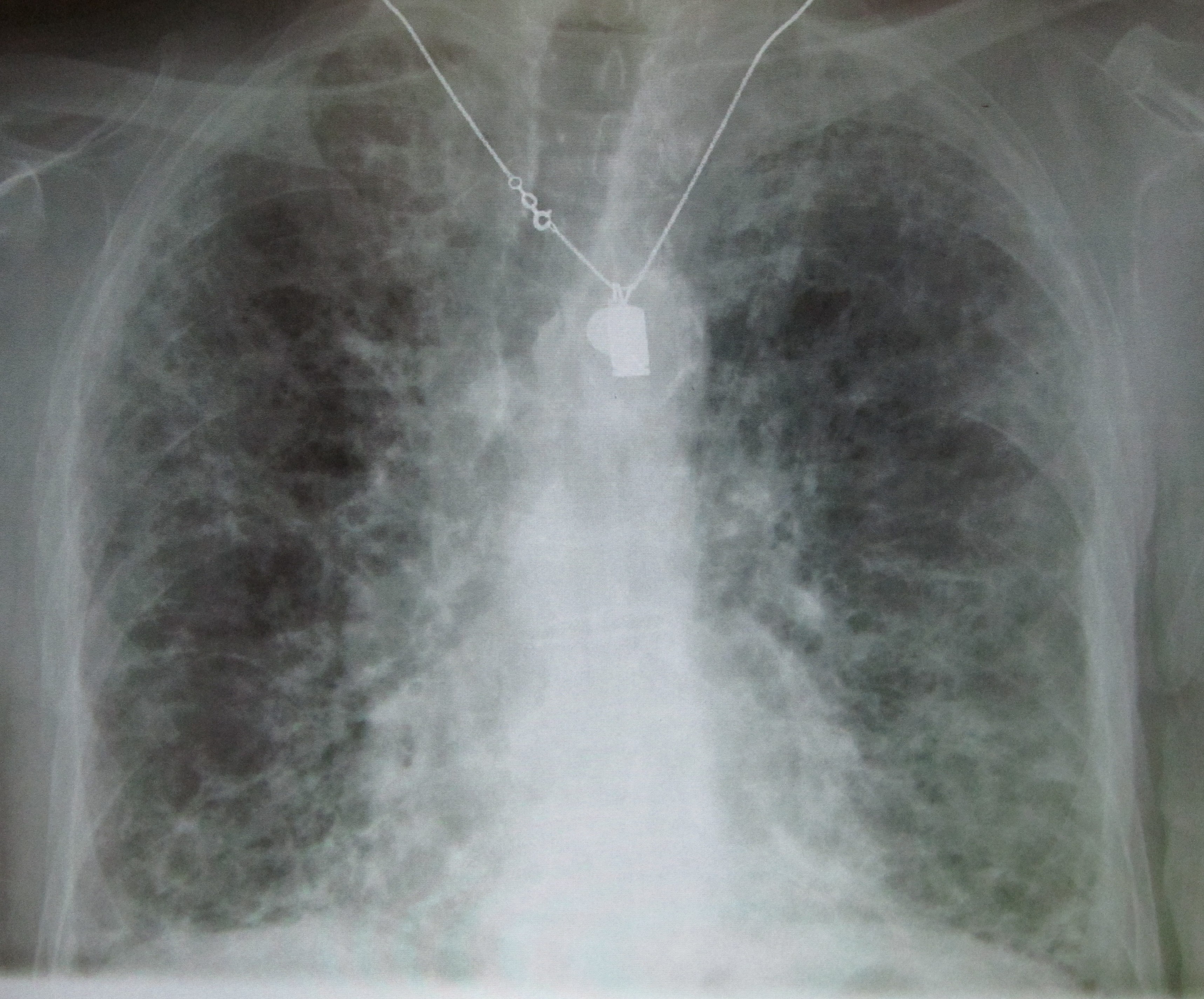
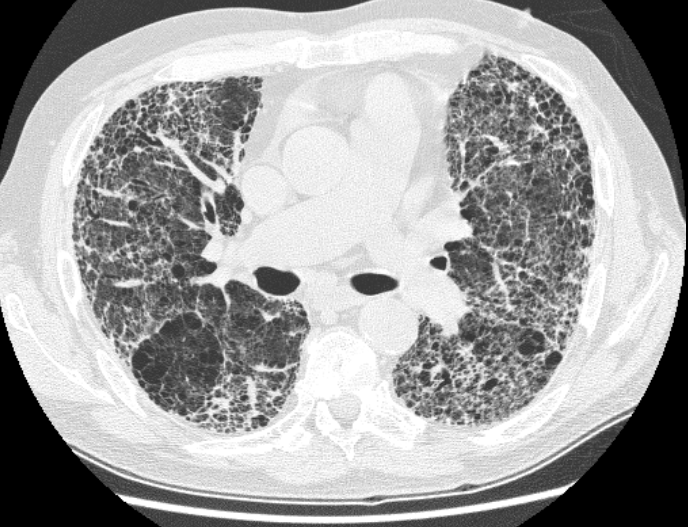
A chest X-ray can disclose features which are suggestive of fibrosis. A high resolution CT scan can add further detail.
Lung biopsies obtained by thoracoscopy tend to be more useful than bronchial biopsies in providing an informative histopathological sample, should histopathological evaluation be required. These biopsies are often small wedges of lung. Contrary to the possible belief of some people who send these biopsies they cannot be reliably interpreted without being supplied with the full clincial and radiological context, in conjuction with the results of other investigations.
One of the roles of a lung biopsy is to check for the presence of features that are incongruous with a diagnosis of idiopathic pulmonary fibrosis.
Blood tests also have an important role. Autoantibodies and rheumatoid factor analyses are helpful in excluding connective tissue disorders. As is almost always the case for dyspnoea a full blood count is useful to check both that the patient does not have anaemia which is exacerbating the symptoms or has developed polycythaemia secondary to chronic hypoxia.
Other investigations will proceed as appropriate for excluding tuberculosis or sarcoidosis if either of these diseases is a possible differential diagnosis for the fibrosis.
|
|
A chest X-ray of advanced pulmonary fibrosis
Image courtesy of Wikipedia
|
A CT scan of marked pulmonary fibrosis
Image courtesy of Wikipedia
|
Treatment
Treatment is by corticosteroids. The response rate is considerably better in desquamative interestitial pneumonia than usual interstitial pneumonia (when it may be as low as 12%). Cyclophosphamide may be employed if prednisolone is ineffective. Lung transplantation is a possibility.
The median survival is six years.
Extrinsic Allergic Alveolitis
Extrinsic allergic alveolitis is a form of type three hypersensitivity reaction that is caused by the inhalation of a triggering antigen by a susceptible individual. It is a rare disease although various different antigens and forms have been described.
Farmer's lung is caused by spores from micropolyspora faeni or thermoactinomyces in mould hay.
Bird fancier's lung is due to proteins on the feathers or in the droppings of various birds, including pigeons and pet birds.
Malt workers' lung is produced by aspergillus clavatus in mouldy barley.
Humidifier fever is caused by thermophilic actinomyces.
Bagassosis is due to mouldy sugar cane.
Pathology
The pathological process is that of type three hypersensitivity. Inhalation of the antigen precipitates a chronic inflammatory cell response. The walls of the alveoli and respiratory bronchioles become distended by this infiltrate, although this distention will resolve once the antigen is cleared and the stimulus for the inflammation is removed. Granulomas are found in around 70% of cases.
If the exposure to the antigen is repeated and/or chronic the inflammation can induce fibrosis and the disease progresses to pulmonary fibrosis. In severe cases a honeycomb lung can develop.
Clinical Features
An acute reaction can occur several hours after exposure to the antigen. The patient has a dry cough and dyspnoea that are accompanied by fever, malaise and myalgia. Examination of the lungs at this juncture discloses widespread crackles (because the inflammation and attendant oedema stiffen the airways).
The presentation may be subacute and require weeks to develop. A dry cough and dypsnoea are again the main features. Crackles are heard on auscultation of the lungs.
One of the key aspects of the acute and subacute presentations is the episodic nature of the symptoms and their relationship to exposure to the antigen. Once the exposure is removed the symptoms should resolve and the patients lungs should return to normal.
If the disease becomes chronic then the features are those of pulmonary fibrosis.
Investigations
The identification of possible exposure and the relationship of the symptoms to this exposure in the history is central to the diagnosis and the investigations are often intended to assess the severity of the disease and to confirm the nature of the antigen rather than to make the diagnosis as such.
The chest X-ray may be normal or may disclose patchy or diffuse infiltrates. Lung function tests will be normal or disclose a restrictive pattern.
Skin tests can determine the identity of the precipitating antigen. Serology may also be useful.
A brochoalveolar lavage can demonstrate an increase in the number of CD8 positive T cells. Lung biopsy is seldom necessary.
Treatment
The essence of treatment is to avoid exposure to the offending antigen. Active acute and subacute disease are treated with steroids.
Depending on the causative antigen the patient may be entitled to industrial injuries compensation.