Contents
Introduction
Haemolytic anaemia is the term applied to a group of conditions in which red cells are inappropriately destroyed at an accelerated rate and have a shorter lifespan. It encompasses both congenital and acquired conditions and includes representatives of instrinsic red cell defects and haemoglobinopathies.
Haemolytic anaemias may be further classified as intravascular or extravascular. In extravascular haemolytic anaemia the site of the destruction of the red cells is the spleen (and to a lesser extent the other parts of the reticuloendothelial system, such as the liver) whereas in intravascular haemolytic anaemias the erythrocytes are clobbered in the non-splenic blood vessels.
The types of haemolytic anaemia are as follows.
- Automimmune haemolytic anaemia
- Sickle cell anaemia
- Glucose-6-phophate dehydrogenase deficiency
- Pyruvate kinase deficiency
- Hereditary spherocytosis
- Hereditary elliptocytosis
- Paroxysmal nocturnal haemoglobinuria
- Drugs
- Malaria
- March haemoglobinuria
- Prosthetic heart valves
- Thrombocytic thrombocytopenic purpura
- Haemolytic uraemic syndrome
The location of the haemolysis may be either within the blood vessels (intravascular haemolysis) or outside the vasculature (extravascular haemolysis). Extravascular haemolysis usually happens in the reticulendothelial system.
Pathology
In addition to the anaemia the process of haemolysis can have several other consequences.
The increased rate of destruction of the erythrocytes elevates the workload of the spleen, particularly in extravascular haemolysis. A modest degree of splenomegaly is therefore common.
The fall in the peripheral blood red cell count stimulates the bone marrow to make more erythrocytes and thus the bone marrow exhibits erythroid hyperplasia.
The destruction of the red blood cells increases the quantity of the haem breakdown product bilirubin. This bilirubin must be metabolised and excreted by the liver. At higher levels of haemolysis this bilirubin load can overwhelm the liver's capacity and the unconjugated bilirubin accumulates, resulting in
jaundice.
Even if jaundice does not develop the liver is pumping increased quantities of conjugated bilirubin into the bile and this means more bilirubin is available for the intestinal bacteria to conjugate to urobilinogen. As a result more urobilinogen is reabsorbed and subsequently excreted in the urine, with the consequence that the urine becomes darker.
The increased concentration of bilirubin in the bile can precipitate the formation of
pigment gallstones.
If the haemolysis takes place in the reticuloendothelial system (extravascular haemolysis), the macrophages deal with the free haem. However, if the haemolysis is intravascular these macrophages are not available to process the haem. The presence of the iron in haem and the function of haem as an oxygen carrying molecule means that it can participate in oxidation-reduction reactions and these reactions can be damaging to blood proteins and cells if the free haem is not removed. Therefore, the blood contains a protein called haptoglobin which binds free haem and stops it causing problems. Intravascular haemolysis consumes some of the haptoglobin and as a consequence the concentration of haptoglobin falls.
Nevertheless, despite the efforts of haptoglobin some of the haem relased during intravascular haemolysis will participate in chemical reactions and these include the formation of methhaemalbumin.
Haemolysis also releases iron. This iron is handled by macrophages in extravascular haemolysis but in intravascular haemolysis the haem is filtered by the kidney which removes reasborbs the iron component of the haem and stores it in the renal tubule epithelial cells as haemosiderin. These cells then slough off and enter the urine, taking the haemosiderin with them. Thus, haemosiderinuria is found in intravascular haemolysis but not extravascular haemolysis.
Haem is toxic to the renal tubule epithelial cells. This is not a problem at normal loads of haem but if a sudden surge of intravascular haemolysis occurs the haem can be sufficient to cause significant damage and rarely acute renal failure. This phenomenon is more of a problem if large quantities of myoglobin assail the kidney, as can occur following rhabdomyolsis secondary to trauma.
Investigations
Before the individual types of haemolytic anaemia are discussed it is prudent to address the investigations that assist in the determining that an anaemia is haemolytic in nature.
The first clue may come from the full blood count. The bone marrow responds to the destruction of the red blood cells by releasing reticulocytes, which are the immediate precursor forms of erythrocytes. Reticulocytes are larger than erythrocytes but are counted by the automated blood analysers as erythrocytes for the purposes of determining the MCV. Thus, the anaemia may be macrocytic. Despite confusing the MCV analysis the reticulocytes can nevertheless be detected by the machine and the full blood count will also show a raised level of reticulocytes.
The blood film can disclose distorted erythrocytes or fragments (schistocytes) and possibly as other features that imply a specific disease. If erythrocytes undergo partial damage and lose part of their membrane they become more spherical.
Haemoglobin electrophoresis can reveal sickle cell anaemia although a specific sickle test can also be performed.
The direct Coombs test is employed to detect autoantibodies to red cells. The direct Coombs test involves mixing the patients washed red cells with antibodies to human immunoglobulins. If the patient's erythrocytes are coated with autoantibodies then the anti-human antibodies will react with these autoantibodies and cause the cells to agglutinate.
The indirect Coombs test is reserved for screening blood for transfusions and for detecting anti-rhesus antigens in maternal blood. The patient's serum is mixed with red cells of a known blood group. If the patient's blood contains antibodies against the test red cells, the red cells will agglutinate.
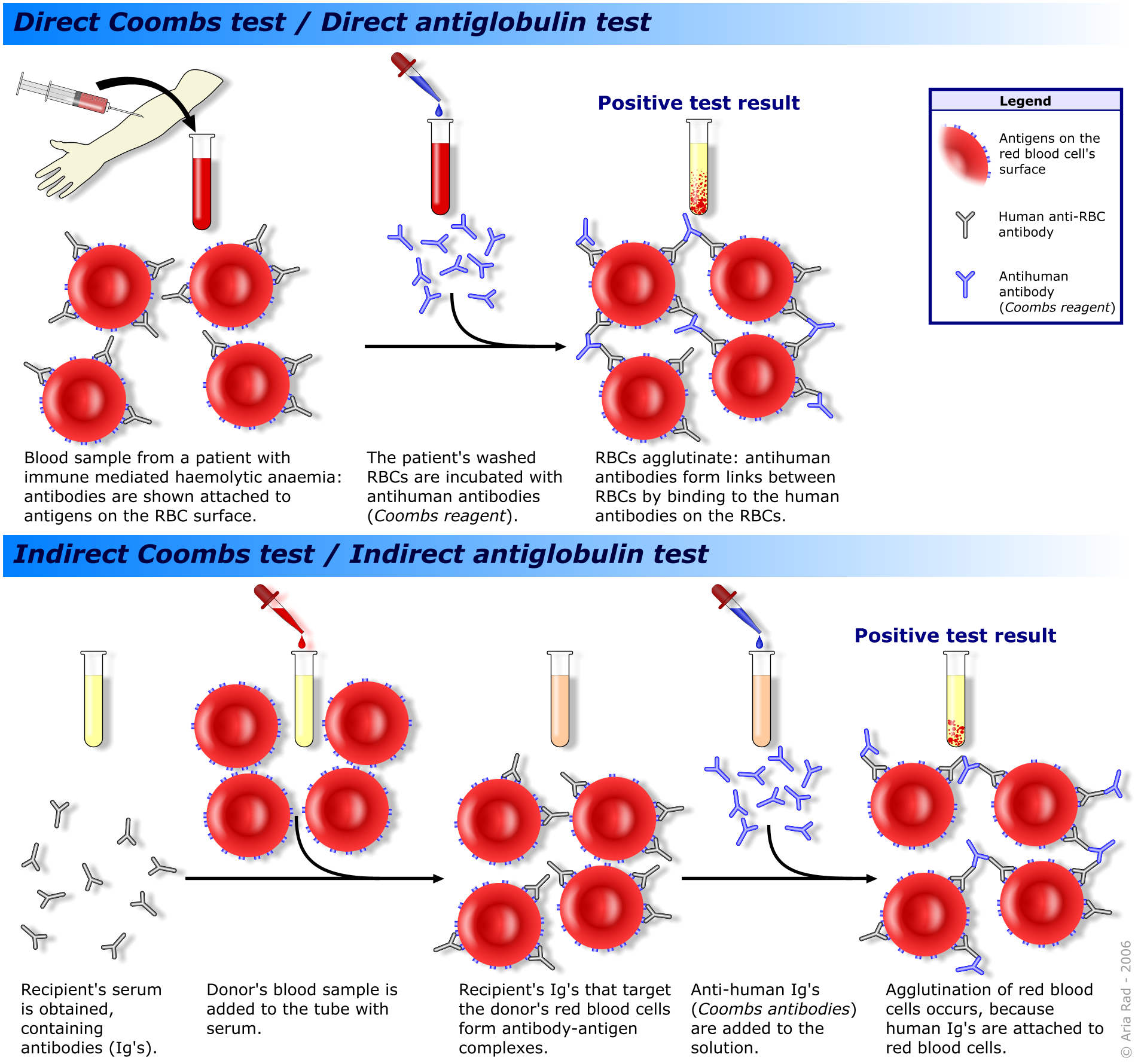
|
The direct and indirect Coombs tests
Diagram courtesy of Wikipedia
|
The presence of haemoglobin in the urine suggests haemolysis; haemosiderin in the urine implies intravascular haemolysis
Levels of methhaemalbumin and haptoglobin can be measured. A low haptoglobin implies intravascular haemolysis.
The levels of vitamin B12 and folate should be checked because the erythroid hyperplasia that tries to oppose the anaemia can consume the reserves of these substances.
Autoimmune Haemolytic Anaemia
Autoimmune haemolytic anaemia is a disease in which the patient manufactures autoantibodies against their own red cells. The antibodies adhere to the red cells and also permit complement to bind. The effect of the binding of antibodies and complement is to render the erythrocytes susceptible to phagocytosis in the reticuloendothelial system and thus the haemolysis tends to be extravascular. However, intravascular haemolysis can also occur, especially in more severe forms of the condition where the autoantibody response is more pronounced.
Autoimmune haemolytic anaemia is classified as warm or cold depending on whether or not the autoantibodies are most active at 37
oC or 4
oC.
Warm autoimmune haemolytic anaemia usually features IgG antibodies. A wide age range can be affected and the disease is slightly more common in females. It is idiopathic in 50% of cases but secondary causes include the following.
- Chronic lymphocytic leukaemia
- Systemic lupus erythematosus
- Rheumatoid arthritis
- Hodgkin lymphoma
- Carcinoma paraneoplastic phenomenon
- Alpha methydopa
Evan's syndrome is the combination of warm autoimmune haemolytic anaemia and autoimmune thrombocytopenia and/or autoimmune neutropenia.
The treatment of autoimmune haemolytic anaemia is steroids.
Cold autoimmune haemolytic anaemia tends to affect the elderly and is caused by IgM antibodies. The haemolysis is more likely to be intravascular than in the warm form and is most likely to affect cold peripheries where the destruction of the red cells can cause temporary vascular occlusion.
The disease is usually idiopathic but can be associated with lymphoma. The treatment is to keep the patient warm.
Acute forms of cold autoimmune haemolytic anaemia can be encountered in mycoplasma infection and Epstein-Barr virus infection (glandular fever / infectious mononucleosis).
Paroxysmal cold haemoglobinuria is a rare form of cold autoimmune haemolytic anaemia in which the patient manufactures an IgG autoantibody (Donath-Landsteiner antibody) directed against the P-complex antigen (the P antigens are red cell proteins that constitute a minor blood group). Patients suffer from bouts of haemolysis that are triggered by exposure to cold. The haemolysis is intravascular and can cause abdominal pain, headache, back pain and peripheral cyanosis. Various infections can trigger the disease and include measles, mumps, chickenpox and congenital syphilis.
Glucose-6-phosphate Dehydrogenase Deficiency
Glucose-6-phosphate dehydrogenase deficiency (G6PD or G6PDD) is an X-linked recessive form of haemolytic anaemia which also falls into the intrinsic red cell defect category of anaemia. The gene is located at Xq28.
The condition affects around 200 million people worldwide. The prevalence is high in Africa, south Europe, the Middle East, Asia and Oceania.
Although the disease is X-linked, recessive females can still be affected by virtue of the phenomenon of
lyonisation, albeit that they manifest the disease less severely than hemizygous males.
Despite the harmful aspects of the condition its prevalence remains high because sufferers have some resistance to falciparum malaria and in overall evolutionary terms this conferred a net benefit.
Pathology
G6PD converts glucose-6-phosphate to 6-phosphonogluconate. This is the entry step into the hexose monophosphate shunt of glycolysis. This shunt allows NADPH to be generated, which in turn is used to generate glutathione. Glutathione helps to protect cells against oxidative stress. As erythrocytes lack mitochondria their metabolic pathways and alternative ways of generating NADPH are somewhat limited and the loss of G6PD renders them susceptible to damage by oxidation.
Under normal circumstances the erythrocytes muddle along and many patients are not permanently anaemic. Instead, anaemia only arises if they are confronted with a challenge which places their erythrocytes under attack from oxidising agents. Such events include infection, anti-malarial drugs, ciprofloxacin, nitrofurantoin, daponse, high dose aspirin, L-DOPA and fava beans. The last of these agents gives the disease one of its other names, favism.
The disease has been subdivided into four types.
Class one has enzyme levels that are less than 10% of normal. These are the patients who do have a chronic anaemia.
Class two exhibits enzymes levels that are also less than 10% of normal but the patients tend only to develop acute attacks of haemolytic anaemia if exposed to a precipitant. Class two is caused by different mutations from class one.
Class three has a similar presentation to class two but the enzyme levels are between 10 and 50% of normal.
Class four disease features patients who do not develop the anaemia. The enzyme levels are no less than 60% of normal, which gives an indication of the reserve capacity of the enzyme.
Clinical Features
The class two and three forms of the disease manifest the typical presentation of acute attacks of haemolysis after exposure to triggering factors. The haemolysis is intravascular. The haemolysis is accompanied by malaise, abdominal pain, back pain, jaundice and dark urine. There can also be fever, tachycardia and splenomegaly.
The class one type of the condition causes a chronic anaemia and possibly jaundice. Almost all patients who have the class one form are male.
Treatment
Acute attacks of haemolysis require removal of the precipitating factor. Blood transfusion is only occasionally necessary. Rarely the patient may need haemodialysis to clear the trigger from the blood.
Chronic anaemia requires supplementation of folate and vitamin B12. Severe cases may benefit from splenectomy.
Pyruvate Kinase Deficiency
Pyruvate kinase deficiency is an autosomal recessive intrinsic red cell defect. Pyruvate kinase is a key metabolic enzyme for the generation of ATP by erythrocytes (other cells are also quite fond of it but have more diverse energy pathways). Thus, the erythrocytes are deficienct in ATP and this crippling of their metabolic pathways causes them to be come rigid. This loss of flexibility is not good for the erythrocytes because it becomes difficult for them to negotiate small capillaries and the splenic sinusoids and they thus become damaged more quickly and removed by the spleen.
The disease causes chronic haemolytic anaemia with attendant splenomegaly. Severe deficiencies present at birth and can produce frontal bossing.
Infection or pregnancy can exacerbate the anaemia.
The red cells accumulate 2-3-diphosphoglycerate. High levels of 2-3-DPG promote the release of oxygen from haemoglobin to the tissues and therefore a given degree of anaemia is better tolerated than that caused by other diseases because the haemoglobin at least surrenders its oxygen more willingly.
Splenectomy can be helpful.
Hereditary Spherocytosis
Hereditary spherocytosis is an autosomal dominant intrinsic red cell defect that is the commonest inherited anaemia in North Europeans where the prevalence is around 1 per 5000.
The genetic defect affects spectrin, which is a structural protein of the erythrocyte membrane. The defective spectrin causes the erythrocyte to lose membrane when it passes through the tribulations of the reticuloendothelial system. Basic geometry forces the cell to become spherical due to the decrease in its surface area to volume ratio. Once this spherocyte form is reached the red cell cannot negotiate the sinusoids of the spleen and is destroyed.
The disease causes a chronic haemolytic anaemia and can present at a variety of ages. Patients are prone to leg ulcers.
The blood film will reveal lots of spherocytes. The erythrocytes also have increased fragility to hypotonic solutions.
The treatment is splenectomy which cuts the red cells some slack.
Hereditary Elliptocytosis
Hereditary elliptocytosis is also an autosomal dominant, intrinsic red cell membrane disorder. Various forms exist and involve the Rhesus antigens, spectrin and other proteins. Only 10% of patients develop anaemia.
Paroxysmal Nocturnal Haemoglobinuria
Paroxysmal nocturnal haemoglobinuria is an acquired intrinsic red cell defect that is caused by an acquired abnormality in haematopoietic stem cells. It is rare and tends to present between the ages of 20 and 40 years. The male : female ratio is equal. The haemolysis is intravascular.
Pathology
The consequence of the defect in the bone marrow stem cell is that the resulting mature blood cells are vulnerable to attack by complement. This susceptibility is due to a loss of the glucosylphosphatidylinositol membrane anchor because the cell lacks the enzyme alpha-1,6-N-acetylglucosaminyltransferase. The function of this chunkily named substance is to anchor proteins in the membrane of the blood cell. Among the proteins that attach to this anchor are decay accelerating factor and membrane inhibitor of reactive lysis (CD59) (these are not the catchiest names but at least they give an idea of what the proteins are). These enzymes help to degrade complement and accelerate the inactivation of C3 convertase and thus protect the cell against the overzealous attentions of complement.
Other deficient membrane proteins include CD67 on T cells, CD74 on mononcytes and CD24 on B lymphocytes. The erythrocytes are also deficient in acetylcholinesterase while neutrophils lack alkaline phosphatase.
Although the name of the disease and its location in textbooks focuses on the effect on erythrocytes, none of the haematopoietic cells is immune. Thrombocytopenia and/or neutropenia can occur and some patients may have a pancytopenia. Progression to
aplastic anaemia may supervene.
Despite the possible thrombocytopenia, patients tend to be hypercoaguable because the complement that coats the erythrocytes can trigger the clotting system.
Clinical Features
The haemolysis is intravascular.
The onset of the disease is usually insidious and the presentation is with lethargy and exertional dyspnoea. There can be jaundice and the intermittent generation of dark urine, often the urine passed first thing in the morning (hence the name paroxysmal nocturnal haemoglobinuria).
Some patients have attacks of abdominal pain of varying severity. These may be due to thrombosis of the small vessels of the portal system.
Thromboembolic phenomena can develop and include deep vein thrombosis, portal vein thrombosis and Budd-Chiari syndrome.
A susceptibility to infection may be present.
Specific investigations include the Ham's test which exposes erythrocytes to a solution at pH 6.4: normal erythrocytes can handle this but those of paroxysmal nocturnal haemoglobinuria cannot. The concentrations of neutrophil alkakine phosphatase and erythrocyte acetylcholinesterase can be measured.
Drug-induced Haemolytic Anaemia
A variety of drugs can cause a haemolytic anaemia. Some act by altering the appearance of erythrocyte surface proteins and making them look foreign to the immune system. Penicillins and cephalosporins are examples of drugs that use this mechanism. The haemolysis in this form is extravascular.
Other drugs, such as sulphonylureas, quinine, rifampicin and digoxin, induce the formation of immune complexes. The immune complexes activate complement and the erythrocytes can be destroyed as bystanders. The antibodies of the immune complexes may act against the free drug, or drug bound to plasma proteins or drug bound to cell membranes. The haemolysis is intravascular and acute. The patient is often systemically unwell and the haemolysis may be of sufficent magnitude to cause acute renal failure.
A few drugs manage to persuade the immune system to make antibodies against the normal components of the red cell membrane. Alpha methyldopa, L-DOPA and mefenamic acid are the main agents that have achieved this notoriety. The haemolysis is extravascular and as with other forms of drug-induced haemolytic anaemia stops quickly once the drug is removed.
Mechanical Haemolytic Anaemia
Mechanical causes of haemolytic anaemia are rare but include march haemoglobinuria and prosthetic heart valves. The former condition affects some people who walk for long distances. The erythrocytes have the hell beaten out of them when they are in vessels of the soles of the feet while those feet are pounding the ground. Mechanical heart valves can place stresses on the erythrocytes and mash them up.