Introduction
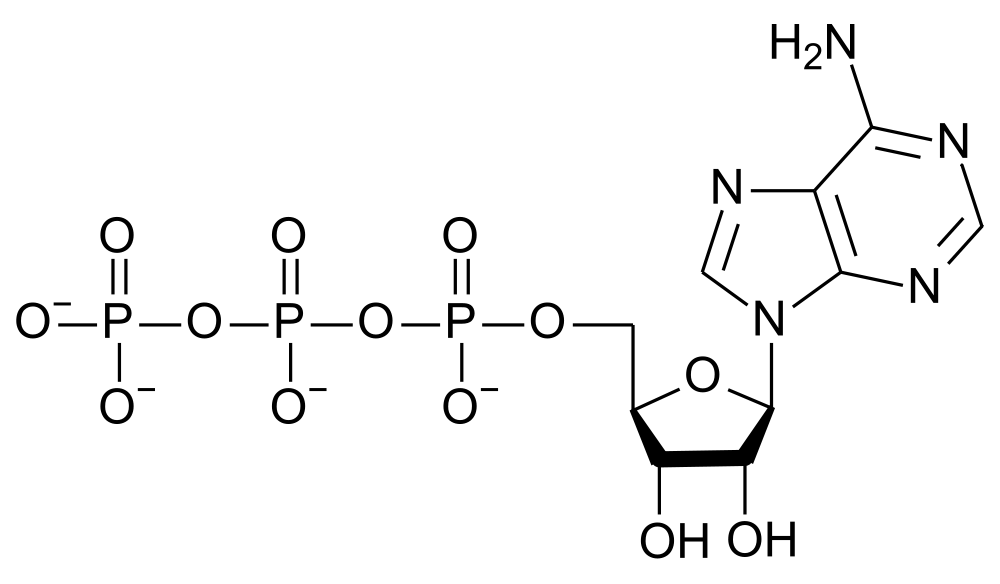
The transduction of energy is a fundamental aspect of human metabolism. Much of the energy liberated from nutrients is stored in the nucleotide molecule adenosine triphosphate (ATP). The three phosphate groups of the ATP molecule are the repository of the energy. The conversion of adenosine triphosphate to adenosine diphosphate (ADP) makes this stored energy available to numerous reactions such as muscle contraction. In addition, the function of many proteins is regulated by phosphorlylating serine, threonine or tyrosine amino acids.
|
|
Adenosine triphosphate
|
The other nucleotide triphosphates have a much more minor role in energy metabolism.
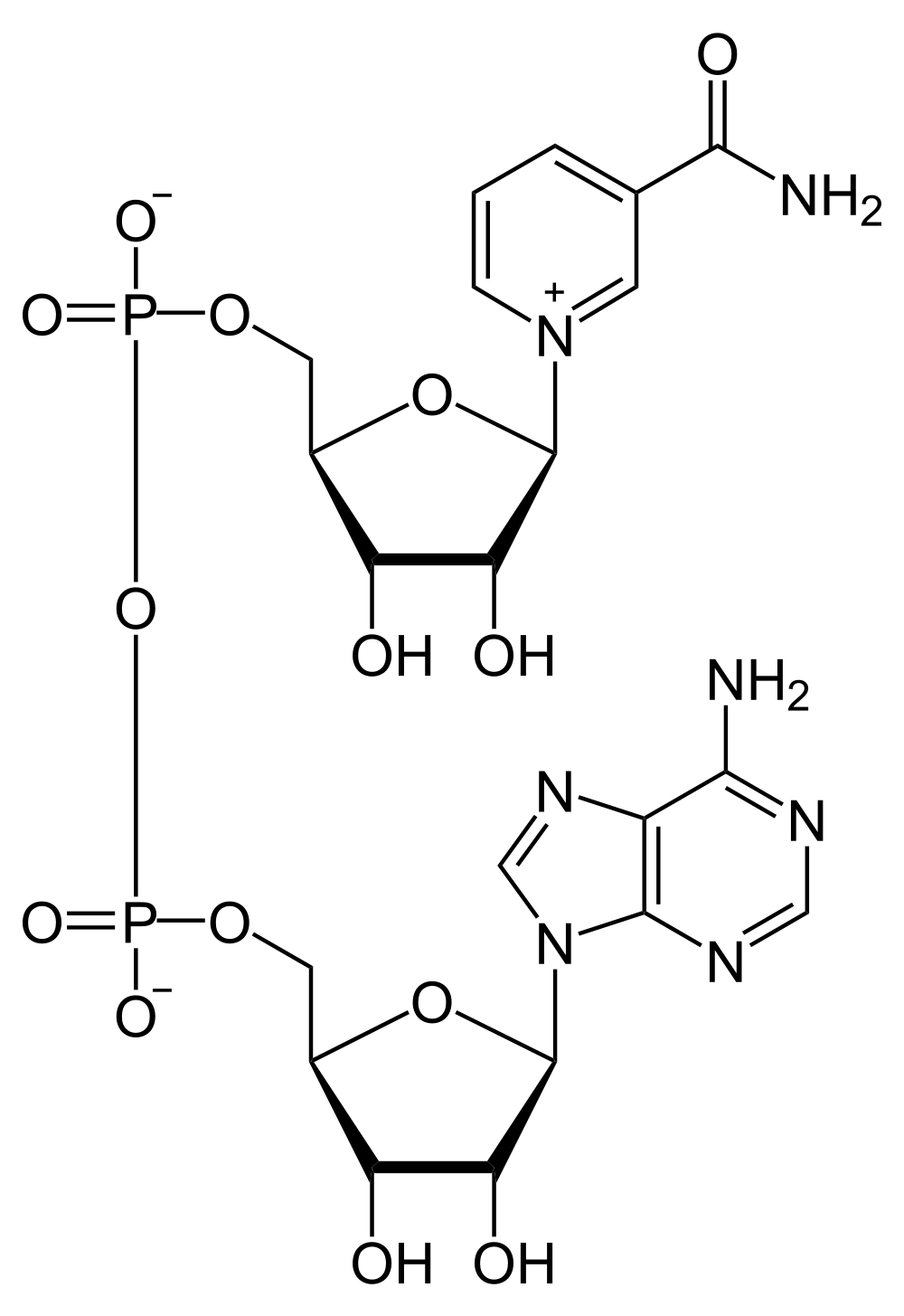
Another molecule that frequently features in many metabolic pathways is nicotinamide adenine dinucleotide (NADH or NAD+). NAD+ is able to receive and transport electrons and is therefore important in many oxidation-reduction processes. Nicotinamide adenine dinucleotide disphosphate (NADPH or NADP+) is a very closely related molecule that differs only be the presence of an extra phosphate group. Like NADH, NADPH participates in oxidation-reduction reactions.
|

|
|
Nicotinamide adenine dinucleotide
|
Conversion between NAD+ (left) and NADH (right)
|
The main substrates that the body uses to liberate energy are carbohydrates (particularly glucose) and fatty acids. Amino acids can be employed in energy generation but as their main function is to form the building blocks of proteins catabolising them in energy production disrupts protein synthesis and therefore tends to be used only as a last resort.
Mitochondria
Mitochondria are intracellular organelles that specialise in energy production. It is believed that mitochondria originally arose from bacteria which formed a symbiotic relationship with early eukaryotic cells. This may explain why a few mitochondrial proteins are still encoded on special mitochondria DNA which is located in the mitochondria and has its own intramitochondrial ribosomes (the majority of mitochondrial proteins are nevertheless encoded in nuclear DNA).
Much of a cell's ATP synthesis occurs in the mitochondria. They are the site at which the Krebs cycle and beta-oxidation of fatty acids takes place.
Mitochondria possess two membranes, an outer and an inner. The outer membrane forms the boundary of the mitochondrion but is freely permeable to small molecules (up to a relative molecular mass of 5000). The inner mitochondrial membrane is far less permeable than the outer and has folds known as cristae which increase the surface area of the membrane. The inner membrane encloses the matrix of the mitochondrion. Many of the special mitochondrial metabolic reactions occur in the matrix.
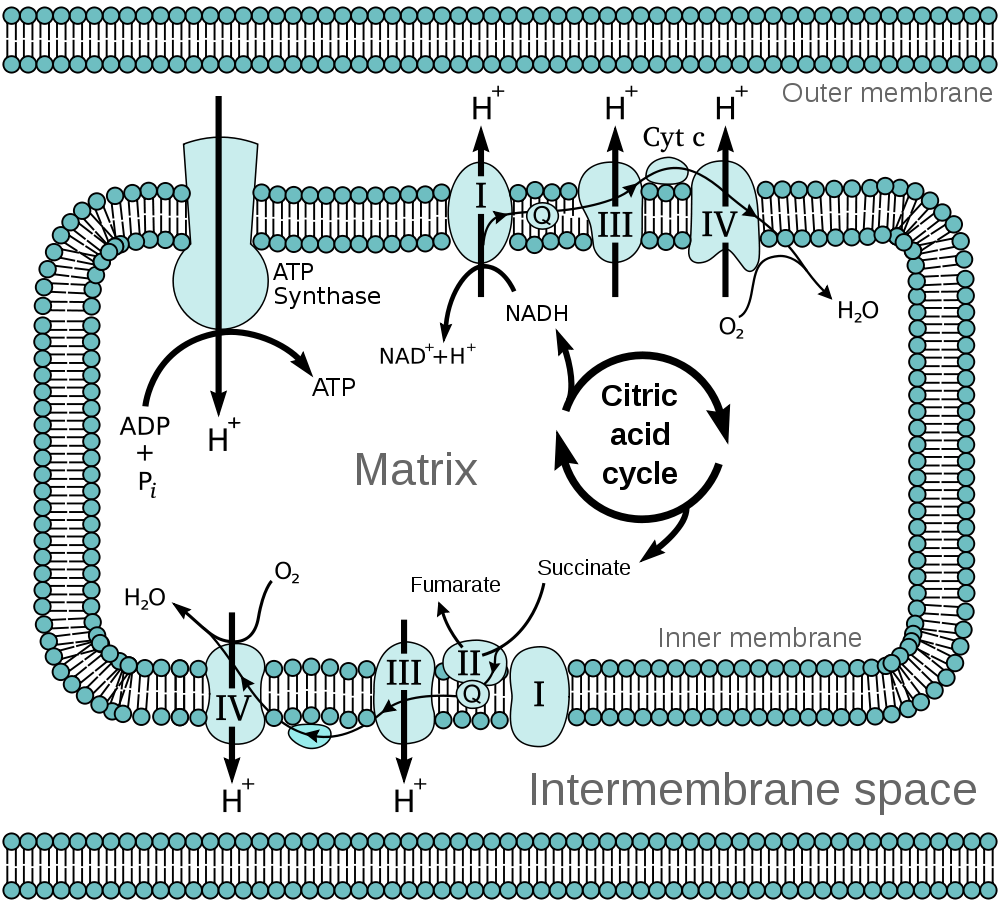
The inner membrane houses the
mitochondrial respiratory chain. This is integral to the process of ATP generation by
oxidative phosphorylation. Oxidative phosphorylation is the process by which oxidation reactions are coupled to the synthesis of ATP. The NADH and NADPH derived from various metabolic reactions in the mitochondrial matrix interact with the proteins of the mitochondrial respiratory chain and result in hydrogen ions (protons) being pumped out of the matrix into the space between the inner and outer mitochondrial membranes. This pumping of protons produces an electrochemical gradient that drives the protons back into the matrix. The re-entry of the protons occurs through the enzyme ATP synthetase. The flow of protons through ATP synthetase causes it to rotate and this rotational energy is transduced into the formation of ATP.
The mitochondrial respiratory chain (also known as the electron transport chain) is composed of four complexes. These feature various proteins and coenzymes. These include cytochromes, which are haem-containing proteins that use iron to carry electrons and coenzyme Q, which can transport both electrons and protons. Complex II of the chain is the succinate dehydrogenase enzyme complex. This enzyme is also part of the
citric acid cycle and uses the electron carrier flavin adenine dinucleotide (FAD/FADH). As well as being an integral cofactor for succinate dehydrogenase, FAD also assists other enzymes that require oxidation-reduction support.
Mitochondria are not involved solely in energy generation. They also serve as a store of intracellular calcium, have a role in apoptosis and assist in the synthesis of steroids. In addition they participate in the production of heat if their proton pumping is uncoupled from ATP synthesis: the energy that would have been used in the creation of ATP is released as heat
|
|
The mitochondrial respiratory chain
|
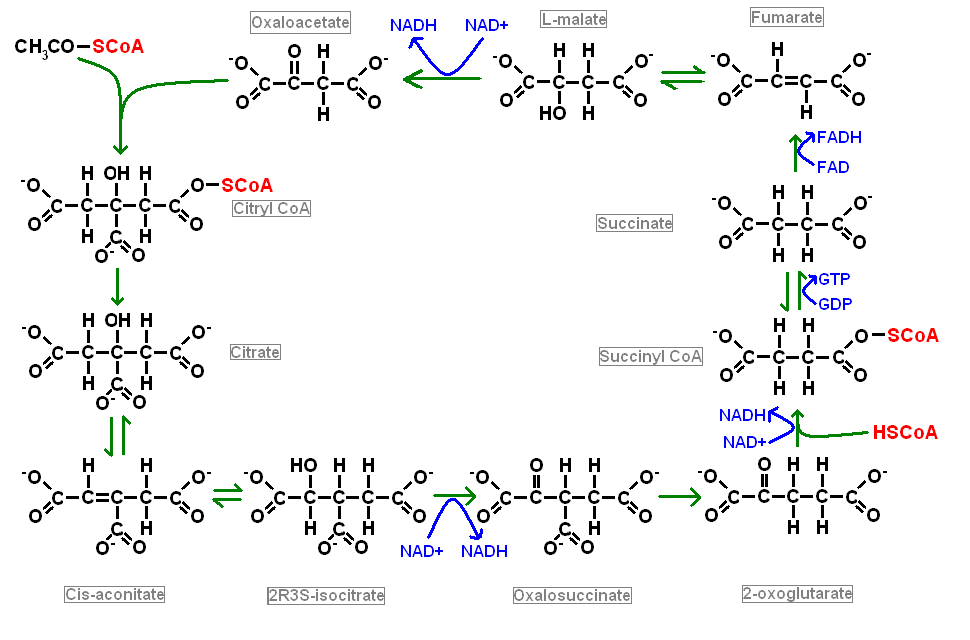
Citric Acid Cycle
The citric acid cycle is also known as the Krebs cycle and the tricarboxylic acid cycle and is an aerobic process. It happens in the matrix of mitochondria and is the main provider of electrons to the electron transport chain. Various metabolic pathways can feed into the citric acid cycle provided that they generate acetyl CoA. The acetyl group of acetyl CoA contains two carbon atoms. The transfer of the acetyl group from acetyl CoA to the the molecule oxaloacetate, which has four carbon atoms, yields citric acid, which possesses six carbon atoms. Citric acid undergoes a series of metabolic conversions into assorted intermediate forms before ending up back as oxaloacetate. Another molecule of acetyl Co donates it acetyl group to this molecule of oxaloacetate and the cycle spins around once more. The carbon atoms that are lost from citric acid in its conversion to oxaloacetate are removed as carbon dioxide.
|
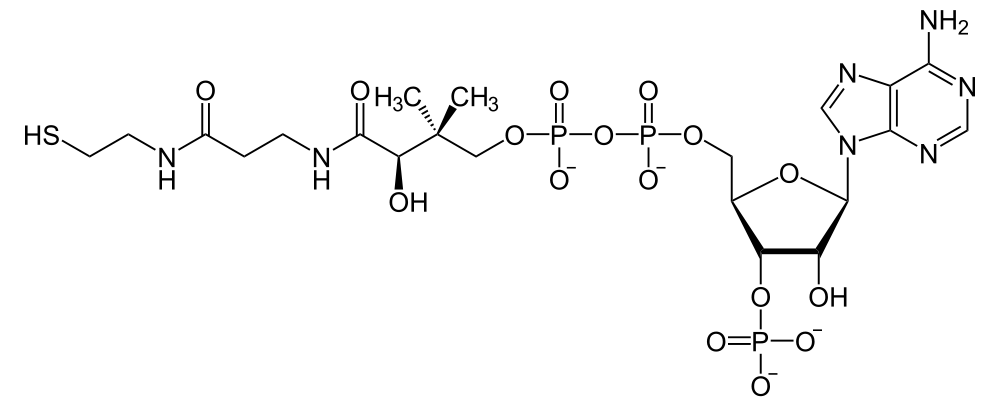
Coenzyme A
Coenzyme A's speciality is carrying small chains of carbon atoms in such a way that makes chemical bond energy available when the carbon atoms are transferred to a target molecule. The SH thiol group at one end of the molecule is the binding site of the carried carbon chains. In the case of acetyl CoA, the attached group is CH3CO.
|
Three reactions in the cycle generate a molecule of NADH which then moseys off to the mitochondrial respiratory chain to help in the production of ATP. In addition, the conversion of succinate to fumarate is catalysed directly by one of the components of the mitochondrial respiratory chain and so more electrons are fed in at this stage. Furthermore the production of succinate from succinyl CoA drives the creation of a molecule of GTP.
Glycolysis, beta-oxidation of fatty acids and the catabolism of amino acids all feed into the citric acid cycle via the acetyl CoA entry point.
|
|
The citric acid cycle
|
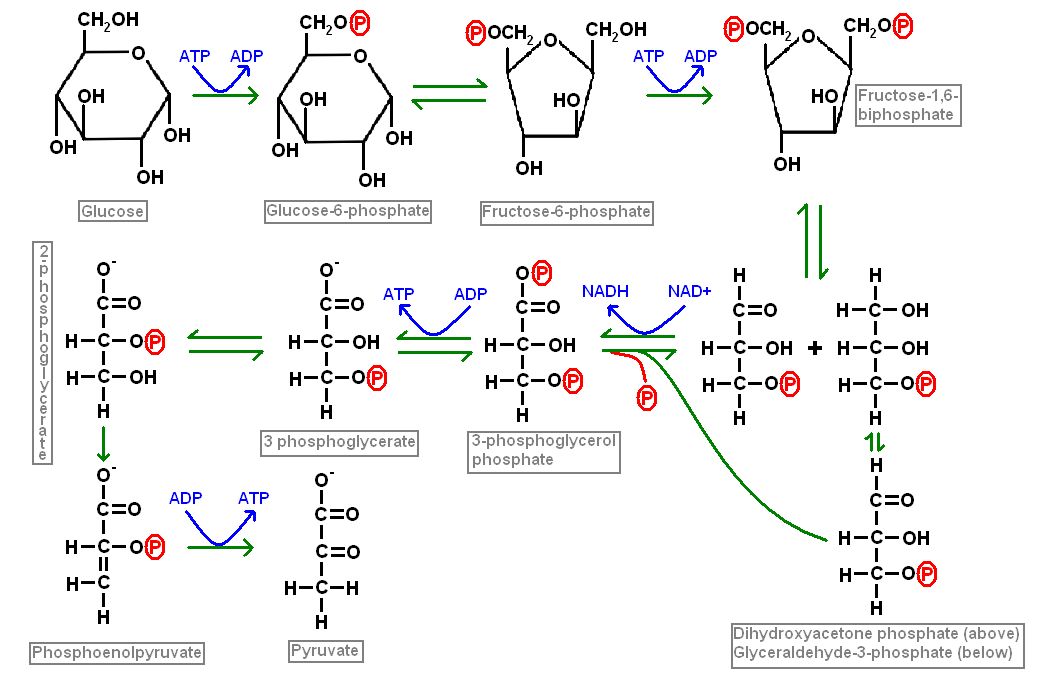
Glycolysis
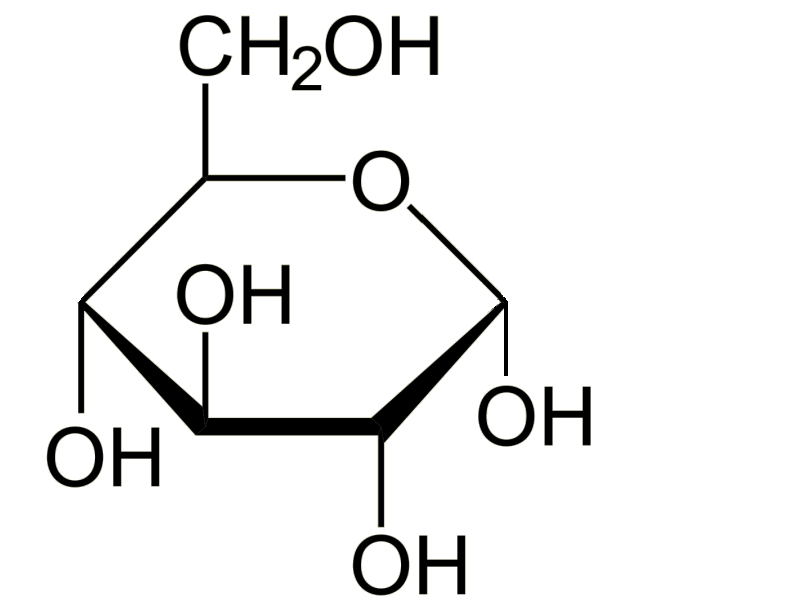
Glycolysis is the process by which glucose is converted in a series of steps into pyruvate. This conversion yields two molecules of ATP and two of NADH per molecule of glucose. Each molecule of glucose generates two molecules of pyruvate. Each molecule of pyruvate can drive one round of the citric acid cycle and thus one molecule of glucose can spin the Krebs cycle round twice.
|
|
Glucose
|
Other carbohydrates, such as galactose and fructose, can enter the glycolytic pathway at different stages through intermediate forms.
Glycolysis is an anaerobic process and does not require oxygen. However, oxidative phosphorylation which utilises the pyruvate generated by glycolysis is dependent upon the availability of oxygen. Pyruvate can be convered into lactate via lactate dehydrogenase (and vice versa) and thus a cell which is forced to depend upon a glycolytic activity without oxidative phosphorylation will generate large quantities of lactic acid.
Glycolysis (glucose to pyruvate) liberates far less ATP than the citric acid cycle. However, glycolysis does produce pyruvate molecules, which in turn allow acetyl CoA to be formed and it is therefore a very important mechanism for feeding substrates into the citric acid cycle. Overall, once a molecule of glucose has passed through glycolysis and the citric acid cycle its catabolism will yield 30 to 36 molecules of ATP.
Glycolysis takes place in the cytoplasm of the cell, not the mitochondria.
|
|
Glycolysis
|
The
hexose monophosphate shunt is a sidearm of the glycolytic pathway that is used to generate NADPH and five carbon ring sugars such as ribose. Its function therefore includes an important anabolic component.
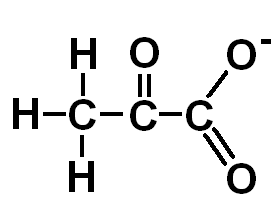
Pyruvate
Pyruvate is the end point of glycolysis and is well suited to be the donor of an acetyl group to coenzyme A to yield acetyl CoA. The creation of acetyl CoA from CoA and pyruvate is performed by the enzyme complex pyruvate dehydrogenase. This reaction occurs in the mitochondria.
Pyruvate can also function as a substrate for the formation of glucose in the process of gluconeogenesis. Furthermore, pyruvate can be converted into alanine (glutamate donates the nitrogen group).
|
|
Pyruvate
|
Beta oxidation
Beta oxidation is the process by which acetyl CoA is generated from fatty acids. It is undertaken by mitochondria. However, before the mitochondria can process the fatty acid, the fatty acid is first complexed with coenzyme A to generate acyl CoA, which is coenzyme A stuck on the end of a fatty acid chain. This process takes place in the cytoplasm and requires ATP. The acyl CoA is bound to carnitine to allow it to be transported into the mitochondria.
Once the acyl CoA has been moved to the matrix of the mitochondria it can participate in beta oxidation. Beta oxidation has four steps. The fatty acid is dehydrogenated, hydrated and oxidated before underoging thiolysis. This last step generates acetyl CoA and leaves the fatty acid chain two carbon atoms shorter than it was at the start of the round of beta oxidation. However, this shortened fatty acid retains a Co A group at the end of its chain and so can enter into another round of beta oxidation and keep re-entering the cycle until the chain has been exhausted. Each round of beta oxidation yields one molecule of NADH and one molecule of FADH, plus all the benefits of the subsequent citric acid cycle that will be applied to the molecule of acetyl CoA that has also been generated. Given the length of most fatty acid chains, their utility as energy stores is considerable.
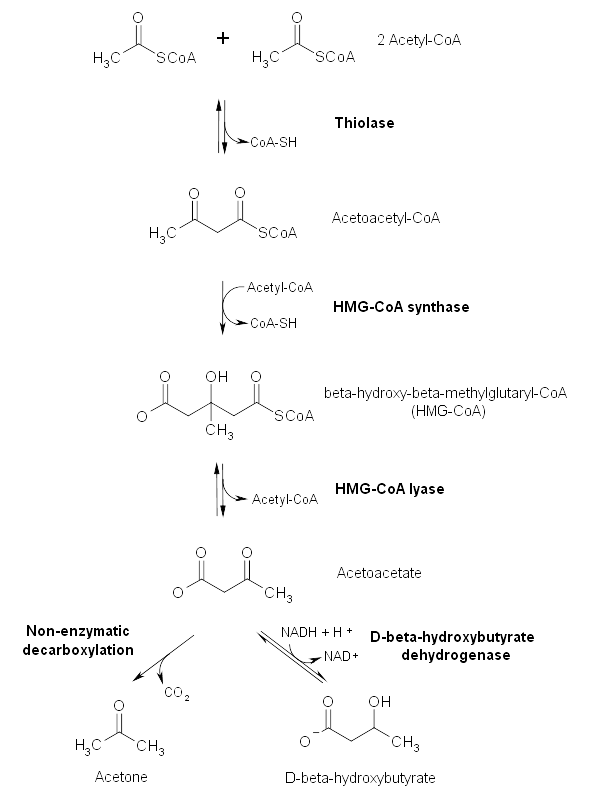
Ketone Bodies
Ketone bodies are derived from fatty acids. Three ketone bodies are employed by the body, acetone, acetoacetic acid and 3-hydroxybutyrate (also known as beta hydroxybutyric acid). The last of these is technically not a ketone and it is unclear why any of the three require the appelation body.
Ketogenesis occurs in the mitochondria of hepatocytes when levels of blood glucose are low. It is inhibited by insulin.
Ketone bodies can be used for the generation of ATP. The principle consumer of ketone bodies is the brain, although the renal cortex and skeletal muscle can also employ them. Ketone bodies are not the brain's preferred energy substrate; neurones are very fond of glucose but if the concentration of blood glucose is low, the brain can switch to ketone bodies (and as importantly cannot really make much use of anything else). The utilisation of ketone bodies for energy is a phenomenon that occurs in fasted individuals and is in effect a back-up plan.
The involvement of ketone bodies in energy production is due to their ability to combine with coenzyme A to generate acetyl CoA which can then feed into the citric acid cycle.
As well as its role in energy metabolism, 3-hydroxybutyrate can also chip in to help out with the synthesis of fatty acids by acting as a supplier of carbon atoms. This can take place in various organs, including the brain. The source of these ketone bodies remains the liver.
|
|
Ketogenesis
|
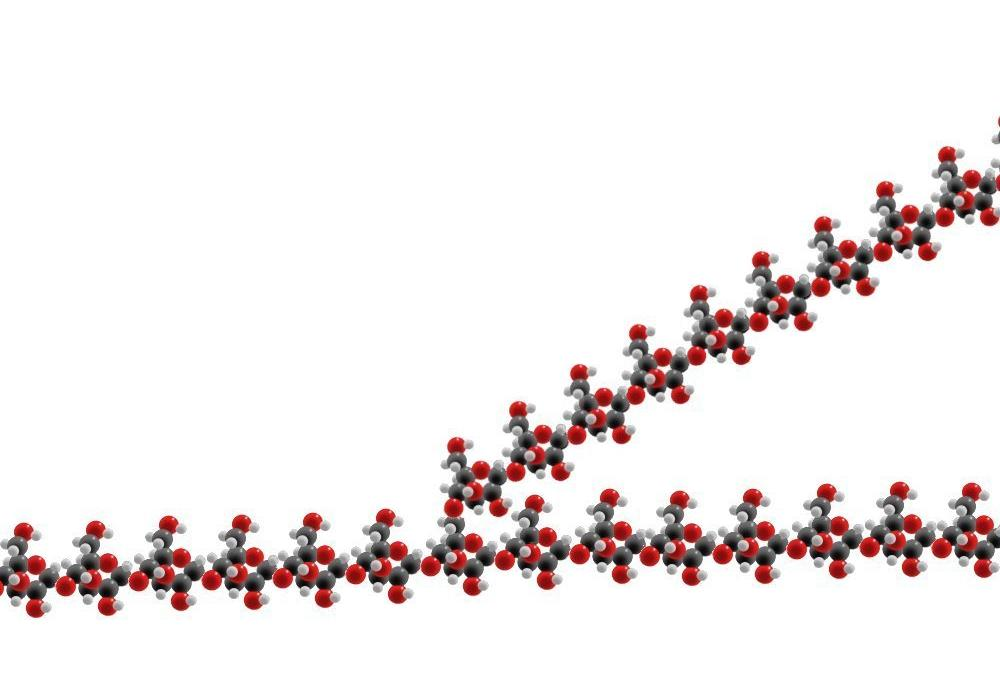
Glycogen
Glycogen is a polymer of glucose. The glucose molecules are linked by the hydoxyl groups attached to the carbon atoms at positions 1 and 4 of the glucose ring. In order for this linking to occur the glucose molecule that is to be added to the glycogen polymer has to be coupled to uridine diphosphate (UDP) at the position 1 carbon atom to give UDP-glucose. This UDP glucose is added to the glycogen molecule and the energy stored in the UDP drives the reaction. The UDP-glucose molecule itself requires the UDP to be donated as uridine triphosphate (UTP); the high energy phosphate group that is sacrificed in the synthesis of UDP-glucose from UTP and glucose makes the reaction energetically favourable. The requirement for a nucleotide triphosphate in the process means that the production of glycogen requires energy. However, this energy is significantly less than that which is liberated from each molecule of glucose when it is released from the glycogen.
Most of the synthesis of glycogen is accomplished by the sensibly named
glycogen synthetase. However, this enzyme can only add UDP-glucose molecules to a pre-existing glycogen molecule.
Glycogenin is the enzyme that makes the initial part of the glycogen molecule.
Glycogen molecules are not simply long linear chains of glucose molecules linked between their positions 1 and 4 carbon atoms. Branches are also present. These are formed by the enzyme
amylo (alpha1-4) to (alpha1-6) transglycosylase which shifts a chain of six or seven glucose molecules at the end of the strand of glycogen onto the position 6 carbon atom of a glucose molecule in the middle of the existing chain. This side branch can then be elongated by glycogen synthetase.
|
|
Glycogen
|
The release of glucose molecules from glycogen is accomplished by
glycogen phosphorlyase. The glucose molecules it frees from the glycogen are phosphorlyated at the position 1 carbon atom (glucose-1-phosphate). Glycogen phosphorlyase cannot handle the first four glucose molecules of a branch. These are instead liberated by a separate debranching system. The enzyme 4-alpha-D-glucanotransferase moves three of the glucose molecules onto another branch of the glucose molecule, where glycogen phosphorlyase can do its thing with them. The last glucose molecule of the four has the 1-6 bond and this is cut by amylo-alpha-1,6-glucosidase.
All of this boils down to the fact that glycogen molecules are great big stores of glucose for cells to use. They require energy to be built and need special enzymes to break them down and make their glucose available to the cell.
Glycogen is synthesised in the liver, muscle and the brain. The muscle and the brain reserve their glycogen for their own use but the liver shares its glycogen with other cells.
Gluconeogenesis
Gluconeogenesis is the creation of glucose from other molecules. The substrates include pyruvate (which can be made from lactate), all the molecules of the citric acid cycle and most amino acids (after their nitrogen group has been removed). Many of the enzymes of glycolysis can catalyse the reaction in either direction so much of gluconeogenesis just involves shifting glycolysis into reverse. THowever, gluconeogenesis takes place only in the liver (and to a small extent in the kidney) and is a response of the body to hypoglycaemia (low blood glucose levels). Most organs are clamouring for glucose and will only be capable of undertaking glycolysis of that glucose, not gluconeogenesis. The hepatocytes of the liver are highly talented chemical processing plants and can derive their substrates for the citric acid cycle from other sources while they are busy beavering away churning out glucose for the rest of the body.
Although most of the stages of glycolysis can operate in both directions without switching enzymes, there are three conversions that require special enzymes to drive in the gluconeogenic direction and this restricts the process of gluconeogenesis to specialised organs.
- Pyruvate to phosphoenol pyruvate
- Fructose-1,6-biphosphate to fructose-6-phosphate
- Glucose-6-phosphate to glucose
All diagrams courtesy of Wikipedia except the diagrams of the citric acid cycle, glycolysis and pyruvate