Contents
Introduction
The purpose of the coagulation cascade is to create a dense network of fibres of fibrinogen that traps erythrocytes and platelets and spans and occludes the breach in the blood vessel. To accomplish this the coagulation cascade is composed of a set of components that interact with each other, both in sequence and with a few feedback loops, to tumble down a metabolic pathway that culminates in the conversion of fibrinogen to fibrin.
Although having multiple components makes the cascade more complex to learn, the cascade system is biologically useful because it allows amplification of an initial, small stimulating signal into a much larger end response (for example, if a cascade has ten components, each of which can activate two copies of the component in the stage below, the activation of one copy of the first component will theoretically generate 512 copies of the end product). A cascade also permits more subtle regulation because there are numerous points at which external factors can impinge on the cascade and modify it.
Most of the clotting factors are proteins that are synthesised in the liver. The principle exception is factor IV, which is simply calcium. Many of the proteins have protease capability, thereby allowing them to cleave their target beneath them in the cascade and activate it by exposing its own protease site. The elements of the coagulation cascade are designated by Roman numerals; an 'a' suffix denotes the active form of the factor.
The Cast
|
Factor
|
Name and notes
|
|
I
|
Fibrinogen
|
|
II
|
Prothrombin. Popular with many other clotting factors. Probably the life and soul of the party.
|
|
III
|
Tissue factor
|
|
IV
|
Calcium. Virtually never known as factor IV.
|
|
V
|
Best friends with factor X.
|
|
VI
|
A case of mistaken identity. Factor VI actually transpired to be just the active form of factor V.
|
|
VII
|
Proconvertin. Popular with factor X.
|
|
VIII
|
Defective in haemophilia A, works as a team with factor IX
|
|
IX
|
Factor VIII's best friend. Deficient in haemophilia B (Christmas disease).
|
|
X
|
Stuart-Prower factor. Works together with factor V in the central role of activating factor II.
|
|
XI
|
Plasma thromboplastin antecedent. Knows how to show factor IX a good time.
|
|
XII
|
Hageman factor
|
|
XIII
|
Fibrin stabilising factor
|
|
No number
|
Prekallikrein. Likes to lavish attention on factor XII, when not attending to its own needs.
|
|
No number
|
High molecular weak kininogen. Cofactor in the activation of factors XI and XII and also prekallikrein.
|
|
No number
|
Von Willebrand Factor. Acts as a minder to factor VIII and helps platelets stick to things.
|
The Diagram
A diagram of the clotting cascade is basically essential to any self-respecting discussion of blood clotting so, courtesy of Paint, an attempt is given below.
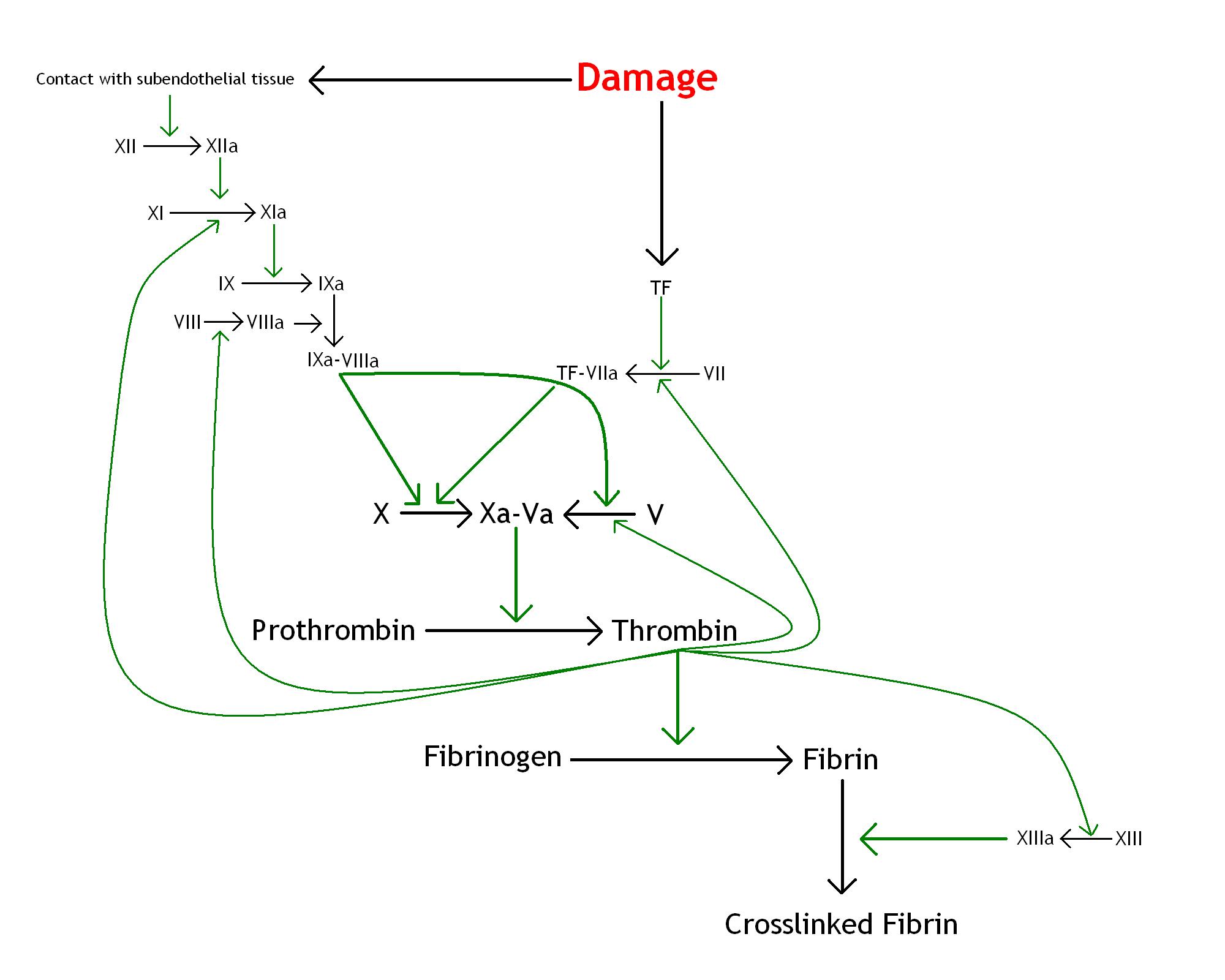
Analysis
Faced with a complex bioloigical process, a student has two choices, assuming that they do not possess an eidetic memory. They can either force the diagram into their heads in the absence of understanding or they can try to comprehend what is going on and make the process of memorisation a bit less brutal.
The goal of the coagulation cascade is to transform an initial modest stimulus into a powerful reaction that generates a cross-linked network of fibrin. Blood cells and platelets become trapped in this meshwork and generate the haemostatic plug which occludes the breach in the blood vessel.
Fibrin (clotting factor Ia) is derived from the precursor fibrinogen (factor I). Cross-linking of fibrinogen is induced by factor XIIIa.
Thrombin (factor IIa) is the agent which converts fibrinogen into fibrin. Thrombin is itself the active form of prothrombin.
The conversion of prothrombin into thrombin is accomplished by activated factor X operating in concert with factor Va.
So far, things have been nice and linear: the factor Xa-Va double act converts prothrombin into thrombin. The thrombin kicks fibrinogen into action and factor XIIIa ties things up. Unfortunately, things become more complicated when the upper reaches of the cascade are explored.
The coagulation cascade could seemingly be triggered by two pathways, the intrinsic (contact activation) and the extrinsic (tissue factor).
The extrinsic pathway utilises tissue factor as the initiating stimulus. The tissue factor activates factor VII, which in turn activates factor X, assisted by calcium. For the purposes of memorisation this has the advantage of being nice and direct.
The intrinsic pathway adds complexity to the diagram. It is triggered when factor XII in the blood is exposed to subendothelial tissues; HMWK and prekallikrein contribute to the fray at this juncture. This contact with subendothelial tissues activates factor XII, which activates factor XI, which in turn activates factor IX. Factor IX then forms a complex with activated factor VIIIa to activate factor X, again with the assistance of calcium.
At this point the instrinsic pathway has slipped in a sleight of hand: factor VIIIa is invoked but no mechanism for activating factor VIII has been stated.
Factor VIII is activated by thrombin. This would seem to generate a chicken and egg paradox. Thrombin activates factor VIII but activated factor VIII appears to be necessary to create the thrombin in the first place.
The explanation is grounded in the relative importance of the extrinsic and intrinsic pathways in initiating clotting. The extrinsic pathway is considered to be the dominant force in the process. The role of the intrinsic pathway is not so much to start the process in the first place but to reinforce it once it has begun. Positive feedback is a key component of the coagulation cascade. Thrombin facilitates the activation of not only factors XI and VIII in the intrinsic pathway but also factor VII in the extrinsic pathway and factor V and factor XIII in the common pathway.
Deficiencies in the upper reaches of the instrinsic pathway (prekallikrein, HMWK, factor XII) tend not to produce clinically apparent defects of coagulation, which would support the contention that the intrinsic pathway is of little significance in starting the process of blood clotting. However, deficiencies of factor VIII or factor IX in the lower part of the intrinsic pathway cause haemophilia A and haemophilia B respectively because these two factors are required by thrombin to generate positive feedback in the cascade.
Further reinforcement of the cascade is achieved by the ability of factors IIa, IXa, Xa, XIIa to activate factor VII. Factor VIIa complexed with tissue factor can autoactivate inactive factor VII.
Thus, a blood vessel is damaged, the extrinsic pathway is activated, the factor Xa-Va duo converts prothrombin into thrombin. The thrombin converts fibrinogen into fibrin which is cross-linked by factor XIIIa which itself had also been activated by thrombin. The thrombin feeds back into various parts of the cascade, including the intrinsic. Within the instrinsic pathway feedback thrombin gees up the effects of exposure of factor XII to subendothelial tissues and a pincer movement is achieved.
Von Willebrand Factor
In all the excitement of endeavouring to elucidate the cascade von Willebrand factor can be overlooked.
Von Willebrand factor (vWF) is a protein that helps the process of clotting along but is not directly part of the coagulation cascade.
Unbound factor VIII is rapidly degraded. As factor VIII has a powerful effect on the coagulation cascade this rapid breakdown helps to limit the action of the cascade once factor VIII is released but it also requires that inactivate factor VIII does not circulate in an unbound form otherwise it will be destroyed before it is needed.
Von Willebrand factor is factor VIII's minder and protects it from degradation when the clotting system is dormant. Thrombin stimulates the release of factor VIII from vWF.
The role of von Willebrand factor is not limited to taking care of factor VIII. Platelets adhere to vWF via the Gp1b-GpV-GPIX complex on their surface. This adhesion is most effective when blood flow is turbulent. Given that vWF attaches to exposed subendothelial tissue, platelets can be channelled to the damaged part of the blood vessel. Furthermore, damaged endothelial cells themselves express vWF.