Contents
Introduction
In the stricter sense of the term, genetic disease applies only to those conditions that are caused by abnormalities of specific genes. However, it is also used more generally to encompass disorders that are due to defects which affect whole chromosomes or parts of chromosomes.
There are several terms which relate to genetic disease that need to be defined.
|
Inherited disease
|
An inherited disease is one which can be passed from a parent to a child via a defect in the genome. There is either a family history of the disease and/or the presence of the abnormal gene can be demonstrated on genetic testing.
Occasionally, an inherited disease will occur in an individual due to a new mutation that either affects one of their parent's gametes, or is acquired by their genome early in embryogenesis.
|
|
Congenital disease
|
A congenital condition is one that is present at birth. It may be caused by an inherited genetic defect, but could also be produced by a problem that occurred during embryogenesis and be neither inherited from the genome of the parents nor transmissible to the offspring of the affected individual. The general use of the term implies a disease which falls under the latter definition.
|
|
Diploid
|
The normal human chromosomal complement consists of twenty-three pairs of chromosomes, yielding a total of forty-six chromosomes. A diploid cell contains these twenty-three pairs. Of the twenty-three pairs, the sex chromosomes form one pair and the other twenty-two pairs are autosomes.
|
|
Haploid
|
A haploid cell contains twenty-three single (unpaired) chromosomes. The only cells that are haploid are the gametes. Haploid gametes are necessary to ensure that when a normal sperm and normal oocyte fuse the resulting zygote has twenty-three pairs of chromosomes, with each pair comprising one maternally derived chromosome and one paternally derived chromosome. The special type of cell division by which haploid gametes are created is known as meiosis.
|
|
Aneuploid
|
An aneuploid cell has an abnormal number of chromosomes that is not an integer multiple of 23.
|
|
Allele
|
With the exception of genes that are located on either the X or Y chromosomes in a male, all nucleated cells possess two copies of each gene. Each copy of a gene is referred to as an allele and can be specified as either the maternal or paternal. The genetic sequence of the two alleles will usually be identical, but different copies can exist, either due to disease or natural variation. (A few genes are represented by more than two alleles, such as the alpha chain of haemoglobin which is duplicated in the genome and has four alleles).
|
|
Homozygous
|
Homozygosity indicates that a person's genome contains the same genetic sequence at both alleles.
|
|
Heterozygous
|
Heterozygosity denotes that the genetic sequences of the two alleles are different (for example one normal and one abnormal).
|
|
Polymorphism
|
In genetics, polymorphism refers to the naturally occuring variation that is found in some genes. A gene is polymorphic if one or more of its variants exist at frequency of at least 5% in the population. The most familiar example is eye colour.
|
Autosomal Dominant
An autosomal dominant disease is one in which the disease will develop if only one of the alleles is abnormal; possession of a normal copy is insufficient to offset the effects of the abnormal gene and the disease will occur in heterozygotes. The offending gene is situated on one of the autosomes. Homozygosity for the abnormal gene may be fatal in utero or lead to a more severe form of the disease.
Autosomal dominant diseases often affect structural proteins. Building a structure in which half of the bricks are malformed does not end well.
Examples of autosomal dominant diseases include Marfan syndrome, achondroplasia and Huntington chorea. A non-pathological examples of an autosomal dominant phenomenon is a cleft chin.
Penetrance is a phenomenon that is encountered in some autosomal dominant conditions and relates to the fact that not all heterozygotes will develop the disease. A condition with a high penetrance is one that affects most people who are heterozygous for the defective allele; a low penetrance means that some heterozygotes may escape the effects of the disease. Anticipation is a related term that reflects the process by which the age of onset becomes earlier and the severity of the disease worse with each successive generation that is affected by it within a particular family. Huntington's chorea shows anticipation. Anticipation is often related to mutations that are caused by repeats of trinucleotide sequences. These sequences are prone to elongation.
Family trees in autosomal dominant conditions show affected individuals in multiple generations. An affected individual will have an affected parent and an unaffected family member will not have any affected children (baring the other parent coincidentally being from a separate affected family). Penetrance can sometimes cause the disease to seem to skip a generation and thus break the third statement.
|
In this example of a family three of an autosomal dominant disorder, the grandmother has the disease. She has passed the condition on to all three of her children, who have in turn transmitted it to three of their seven children. Tranmission has occured from female to female, female to male, male to female and male to male, eliminating X-linked and mitochondrial inheritance as possibilities. The penetrance in this example is 100%.
|
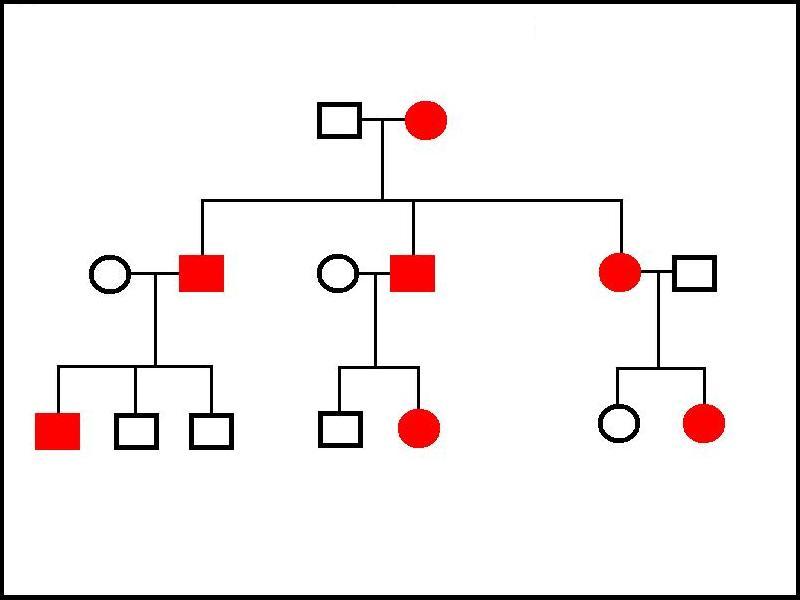
|
Autosomal Recessive
An autosomal recessive disease is one in which the defective gene is located on one of the autosomes and both alleles need to be abnormal for the disease to be manifested. In other words, the disease is not apparent unless the person is homozygous for the mutated gene.
Autosomal recessive diseases tend to involve enzymes. Most enzymes operate with sufficient efficiency that a cell can function normally even if half of the copies of the enzyme it makes are defective.
Examples of autosomal recessive diseases include cystic fibrosis, haemochromatosis, Wilson's disease, homocystinuria and phenylketonuria.
Family trees in autosomal recessive diseases do not show as many affected people as in autosomal dominant conditions. Patients tend to have parents who are not affected but instead are both heterozygous for the gene and are termed carriers. If two carriers have children, one quarter of the offspring will be homozygous for the normal gene, one quarter will be homozygous for the abnormal gene and one half will be heterozygous carriers (there is a fifty percent chance that each parent will pass on the normal allele to each child and a fifty percent chance that they will transmit the defective allele).
A sufferer of the disease is certain to pass the abnormal gene onto their children (assuming the disease does not cause infertility) because their are homozygous for it, but if the other parent does not carry the abnormal gene at all the children will only be carriers; if the other parent is a carrier, the children will have a fifty percent chance of being homozygous for the defective gene and a fifty percent chance of being carriers; if the other parent is also homozygous for the disease, all the children will also be affected.
|
This family tree shows four generations of three families. The great grandparents in the right hand family are both carriers and produce one affected daughter and one carrier daughter. The carrier daughter has three children with a carrier male; one of these children, a son, is affected, the other two children are homozygous for the normal gene. One great grandparent in the left and middle branches of the tree is a carrier and carriers are present in each generation until two of them marry to yield an affected daughter in the fourth generation. The gene displays no gender restriction in transmission. Note how this family tree needs to be more extensive than the tree for the autosomal dominant disease in order to elucidate and demonstrate the nature of the mode of inheritance.
|
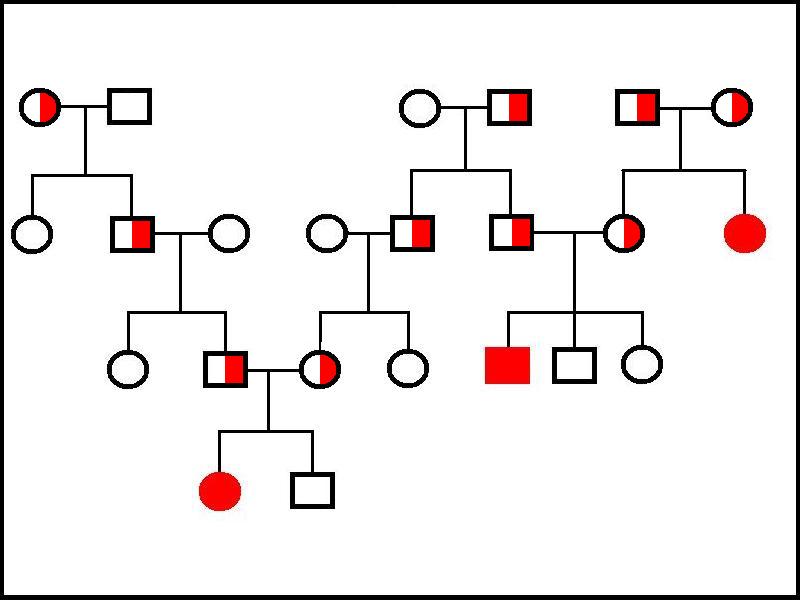
|
X-Linked Diseases
X-linked diseases are caused by genes that are located on the X chromosome. With very few exceptions the diseases are all recessive conditions, which means that heterozygous females will not normally be affected by the disease because their one good copy of the gene is enough to protect them. By contrast if a male has the abnormal gene he will suffer from the disease because he has no spare copy. Contrary to popular belief X-linked recessive diseases can occur in females provided that the female is homozygous for the gene (usually because her mother is a carrier and her father has the condition).
X-linked diseases that behave in a dominant fashion are typically fatal in utero in males. The principle exception is vitamin D resistant hypophosphataemic rickets which is not incompatible with life in males. Aicardi syndrome is a X-linked dominant disease that is only seen in females because male embryos die in utero.
Examples of X-linked recessive conditions include colour blindness, haemophilia A, haemophilia B and Duchenne muscular dystrophy (this is due to a structural protein but violates the general rule given above which states that diseases of structural proteins are dominant).
Y chromsome related diseases have not yet been described, although having hairy ears has been traced to the Y chromosome. The main purpose of the Y chromosome is to commit the embryo to differentiating its immature gonads into testes. The secretion of testosterone by the testes can then take care of most of the other aspects of sexual differentiation into a male.
The genes for X-linked diseases can be transmitted from a mother to either her sons or daughters but a father can only pass the gene onto his daughters; his sons will inherit his Y chromosome not his X chromosome.
|
This family tree illustrates an X-linked disease. In the first generation, an affected brother has a carrier sister. Subsequent tranmission is either carrier status from mother to daughter, carrier status to a daughter of an affected father or disease to a son from a carrier mother. No transmission from father to son occurs. No females express the disease in this family.
|
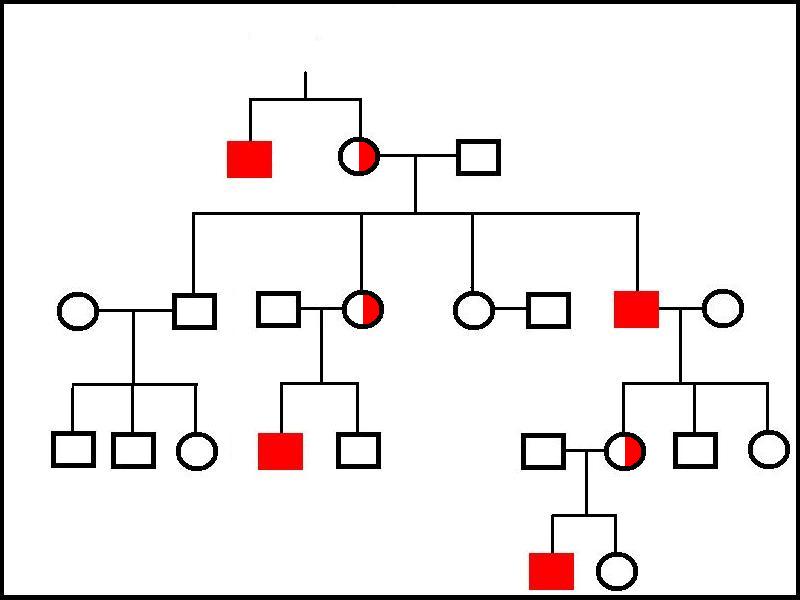
|
Mitochondrial Disease
Mitochondria are believed to have evolved from symbiotic organisms that were incorporated into very primitive single celled or oligocelled organisms in the early stages of evolution. The ability of the ancestral mitochondria to engage in oxidative metabolism and transduce large quantities of energy into a biologically available form made them a valuable acquisition so the arrangement persisted and flourished. These precursors to mitochondria had their own genetic material. As endless aeons wheeled and passed, much of this genetic material became incorporated into the nucleus of the host cell and mechanisms evolved that allowed the host cell to transport the proteins it made from this assimilated mitochondrial DNA back into the mitochondria. However, a few genes have remained within the mitochondrial genome and are transcribed by mitochondrial protein synthesis systems; mitochondria also retain mechanisms for replicating their own DNA. This mitochondrial DNA can be the source of a few rare diseases, such as Leber's hereditary optic neuropathy and MERRF syndrome (myoclonic epilepsy and ragged red fibres).
Mitochondrial diseases typically affect cells that have high aerobic energy requirements and therefore usually involve neurones, skeletal muscle and cardiac muscle. The term mitochondrial myopathy is sometimes employed.
Mitochondrial DNA is passed on by the mother only. While sperm do possess mitochondria, they are situated in the body of the sperm. When a sperm fertilises an ovum the head breaks off and penetrates the ovum alone. Even if a few sperm mitochondria do manage to enter the ovum, their small contribution is overwhelmed by the large array of oocyte mitochondria.
|
This family tree demonstrates the pattern of inheritance of a disorder of mitochondrial DNA. All of the children of an affected mother have the disease. All of her daughters pass the disease to all of their children, while the affected males do not transmit the disease to any of their offspring. No carriers are observed.
|
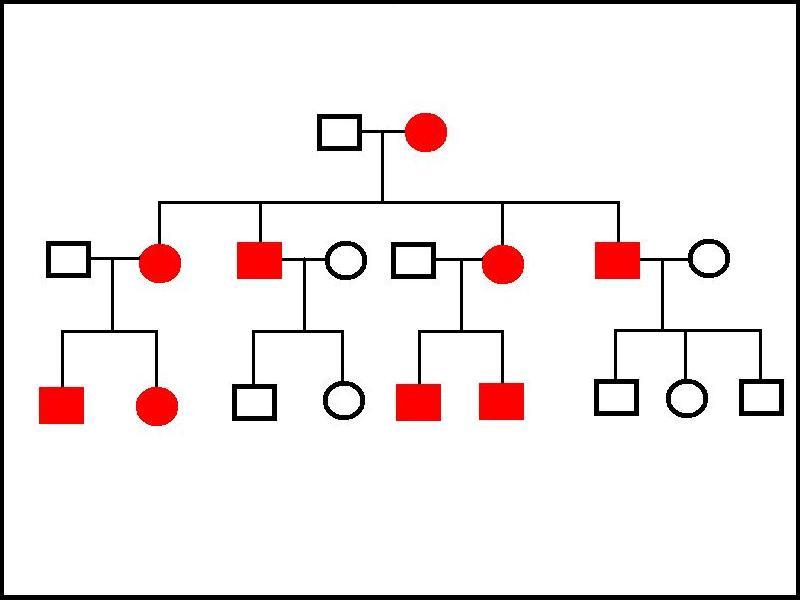
|
Chromosomal Disorders
Genetic abnormalities may not be confined to single genes. In somes diseases the problem exists at the level of the chromosomes. There may be an extra copy of one or more chromosomes, a missing copy, or part of a chromosome may be duplicated or deleted.
Chromosomal diseases arise from an aberrant meiotic division in the gametogenesis of either the father or the mother. Whereas normal meiosis should generate haploid gametes through separation of pairs of chromosomes, defective meiosis may result in one pair of chromosomes failing to separate. If this occurs, one of the gametes will have two copies of the chromosome and the other will have none. If one of these abnormal gametes is used in the generation of a zygote, that zygote will have either three copies (trisomy) or one copy (monosomy) of the affected chromosome.
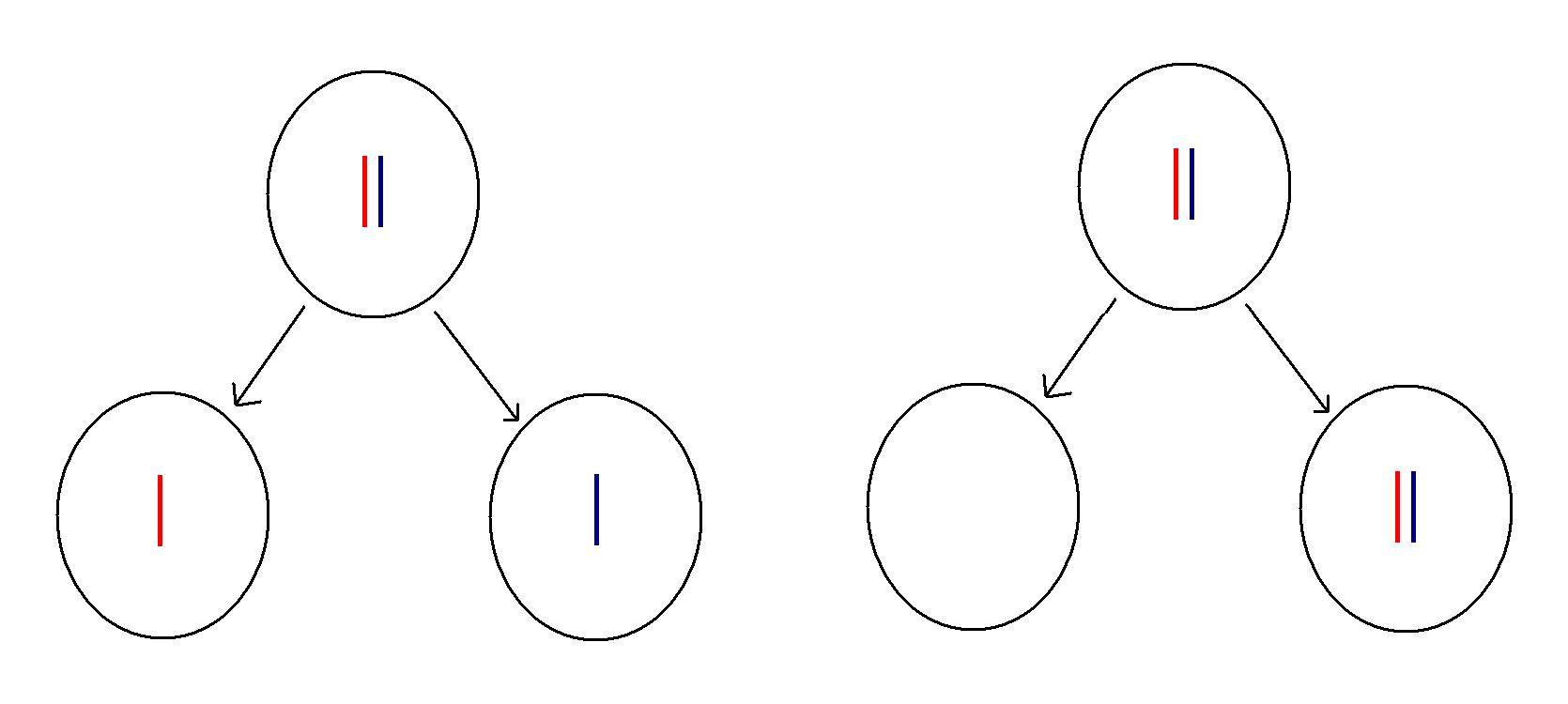
|
|
A normal second stage meiotic division for one pair of chromosomes is shown on the left. The red chromosome is derived from the mother (the grandmother relative to the person who will be created from the resulting gamete) and the blue from the father (grandfather). An abnormal division is depicted on the right. Instead of separating, the two chromosomes remain together, producing two aneuploid gametes.
|
Normal gametes do not need to survive for long. In a short while after their creation they will either fertilise or be fertilised, or they will fail in this task. In the former case they will form a diploid zygote that possesses a normal chromosome complement. In the latter, they will die. Thus, in both situations they only need to survive for a brief interval with a haploid chromosomal complement. However, if an abnormal gamete fertilises or is fertilised, the resulting zygote is faced with a lifetime of aneuploidy. Having only half normal levels of all (or a significant portion) of the genes on a chromosome places considerable demands on a cell. Similarly, having levels that are fifty percent greater than they should be also deranges cell metabolism and function.
Many zygotes that have an aneuploid chromosomal composition are not viable and will result in an early miscarriage. However, there are some chromosomal abnormalities that are compatible with life, albeit that there tend to be multisystem disorders.
The most common and well known chromosomal disorder is Down syndrome. People who have Down syndrome possess an extra copy of chromosome 21 (trisomy 21). Other trisomic diseases are Patau syndrome (trisomy 13) and Edward syndrome (trisomy 18), in both of which survival is for no more than a few months after birth. A small percentage of cases of Down syndrome are due not to an extra copy of the whole of chromosome 21 but are instead caused by an abnormal chromosome 21 that has an extra copy of its long arm attached to it such that the patient has three copies of the long arm of chromosome 21; this is enough to produce the Down syndrome phenotype.
Turner syndrome is an example of a disorder of the sex chromosomes. Affected invididuals have only one sex chromosome (an X) and their karyotype is expressed as 45XO. They are phenotypically female but have developmental abnormalities and are infertile. Klinefelter's syndrome features a 47XXY karyotype. The presence of the Y chromosome is enough to induce male differentiation, although suffers are infertile and have hypogonadism. Rarely, markedly deranged karyotypes such as 48XXXX or 48XXXY are found.
Lyonisation
Females have two X chromosomes while males only have one. If left without adjustment this would mean that either males have fifty percent less X chromosome proteins than they need or females will have double what they require. Both situations should produce disease so some correction needs to be in place.
Cells actually only require one functioning X chromosome. In males their cells only have one, so things are okay as they are. However, females have two, so will receive a harmful double dose of all their X chromosome derived proteins unless something is done. The biological solution to this problem is lyonisation. At a very early stage in the embryogenesis of a female, each cell randomly selects one X chromosome to be deactivated. This spare copy is converted into a Barr body. Patients who have Turner's syndrome do not possess Barr bodies. Individuals who have more than two X chromosomes will have more than one Barr body.
Lyonisation means that females are genetic mosaics. The chromosomal composition of all their cells is not identical because around half have retained one X chromosome while half have kept the other. Occasionally, this can lead to expression of X-linked diseases in a heterozygous woman through bad luck in the choice of X chromosome to be deactivated.
The family tree diagrams modified from the original draft of Pathology in Clinical Practice: 50 Case Studies with the permission of the author.